
研究 up to date
早期発症・急速進行性の経過を取り好塩基性封入体を呈するFUS変異陽性の家族性筋萎縮性側索硬化症
FALS with FUS mutation in Japan with early onset, rapid progress and basophilic inclusion
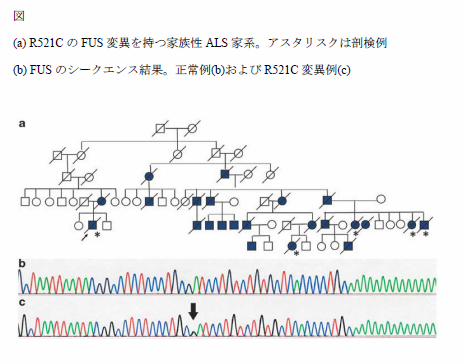
筋萎縮性側索硬化症(Amyotrophic lateral sclerosis;ALS)は運動ニューロンを選択的に侵す難病である。5-10%が家族性であり、家族性のうち20%前後はSOD1の変異が原因となっている。2009年にfused in sarcoma (FUS, 別名translated in liposarcoma: TLS) 遺伝子の異常がアフリカや欧米諸国で家族性ALSの原因遺伝子として報告された。我々は日本人の家族性ALSの大家系において、欧米と共通するR521C位のFUS変異を見出した(図)。
この家系では4世代にわたる46人の家系構成員のうち実に半数にあたる23名が症状を呈しており、浸透率は100%と見積もられる。筋力低下の症状が出現が平均35.3歳であり発症年齢が低い。本家系内の剖検に御協力いただいた症例では顕著な脳幹被蓋部の萎縮が見られ、細胞質の好塩基性封入体の存在が特徴的である。東北大学神経内科では以前より家族性ALSの遺伝子検査を行ってきたが、遺伝子が未確定で常染色体優性遺伝の遺伝形式をとる40家系についてFUS遺伝子をスクリーニングしたところ4つの変異(S513P, K510E, R514S, H517P)をエクソン14および15に見出した。アジア人種においてもFUS変異を持つ家族性ALSは早期発症・急速進行・高浸透率という特徴を持つ。一方我々が見出したS513P変異例は60歳前後発症と緩徐な経過を取る。FUSはTDP43と類似の構造を持ち共にDNA/RNA代謝に重要な働きをすることがわかっており、ALSの病態解明に重要な手掛かりを与えるものと考えられる。好塩基性封入体の病態への関連性の解明が今後の課題である。
{kind=link}
細菌性髄膜炎との鑑別を要した神経ベーチェット病の1例
○祢津昌広 (医学部6年生), 鈴木直輝(臨床神経,48:750―753, 2008 祢津昌広, 鈴木直輝, 水野秀紀, 高井良樹, 三須建郎, 青木正志, 中島一郎, 糸山泰人)
症例は30歳の男性、激しい頭痛, 発熱および軽度の意識混濁を呈し救急搬送された。神経学的所見では髄膜刺激症状陽性、腰椎穿刺にて髄液圧亢進と多核球優位の髄液細胞増多を認めた。MRIでは脳幹部に造影病変を認めた他、粒状の病変を脳内に散在性に認めた(図)。 数年前に神経ベーチェット病(NBD)の診断を受けており、NBDの再発を考えたが、細菌性髄膜炎の合併も当初否定できなかった。メチルプレドニゾロンパルス療法とメロペネム、 バンコマイシンの併用により数時間の単位で症状の改善が得られ、経過から細菌性髄膜炎は否定された。 本症例では細菌感染症で上昇するとされる血清プロカルシトニン値が基準値範囲内であり、両者の早期鑑別に有用と考えられた。
αシヌクレインリン酸化によって誘導される細胞死は小胞体ストレスを介する
Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death.
パーキンソン病は、無動・振戦・固縮を三徴とし、神経内科診療における代表的疾患のひとつである。レボドパの内服が症状を軽減しうるが病態は未解明であり、疾患そのものの進行を抑制しうる治療法はいまだ存在しない。病理学的には中脳黒質のドパミン産生細胞の変性脱落と、レビー小体と呼ばれる構造物が特徴的である。レビー小体はパーキンソン病関連疾患に特異的に現れることから、パーキンソン病における神経変性と密接な関連があるものと考えられている。近年、αシヌクレインという蛋白がレビー小体の中心的構造物であることが判明し、さらにレビー小体中のαシヌクレインは129セリンでリン酸化修飾を受けている事が報告された。また、αシヌクレインの点突然変異、また二重重複・三重重複が常染色体優性遺伝の家族性パーキンソニズムをきたすことからもαシヌクレインはパーキンソン病発症におけるkey moleculeと考えられている。
今回我々はαシヌクレインの神経細胞死における役割について、特に129セリンのリン酸化に着目し実験細胞を用いて検討を行った。まず野生型のαシヌクレイン、または129セリンのリン酸化を阻害する変異S129Aの過剰発現細胞株を作製した。野生型αシヌクレイン過剰発現細胞に低濃度ロテノンを暴露すると、初期におよそ80%のαシヌクレインがリン酸化されていることが明らかとなり、さらに後期にはレビー小体類似の細胞内凝集体と細胞死が観察された。しかし、S129A変異体では、αシヌクレインのリン酸化・細胞内凝集体・細胞死のいずれも生じなかった。細胞内凝集体は、β-COPと呼ばれる小胞体からゴルジへの輸送に関わる蛋白と共在していた(図1)。次いで細胞死に至る経路の検索を行うと、ミトコンドリア障害の出現に先行して著明な小胞体ストレスの誘導が認められた。αシヌクレインが小胞体からゴルジへの輸送を阻害するとの報告があり、本実験の結果と合わせ考察すると、αシヌクレインの小胞体−ゴルジ輸送阻害の結果として輸送途中に凝集体が生じ、また小胞体ストレスが誘導されているものと考えられた(図2)。αシヌクレインのリン酸化はこれらを誘導するために必須であった(図2)。本論文は、パーキンソン病の分子病態において注目されているαシヌクレインのリン酸化と小胞体ストレスの関わりをはじめて明らかにした研究である。
難治性の吃逆・嘔気はNMO再発の前駆症状として重要である
難治性の吃逆・嘔気は、Neuromyelitis optica (NMO) に特徴的な症状と考えられ、我々の以前の検討では、NMOの47例中8例に認められたものの、多発性硬化症130例では1例も認められなかった。これらの症状は、延髄背側の吃逆・嘔吐中枢を含む病変によって生じると推定され、そのMRI画像上の特徴も明らかにした(Misu et al. Neurology 65:1479-82,2005)(図)。 今回、NMOに極めて特異性の高い血清自己抗体である抗アクアポリン4(AQP4)抗体が測定可能になったことから(Takahashi et al. Brain 130:1235-43, 2007)、抗AQP4抗体陽性のNMOおよびHigh-risk syndrome(NMOの部分症と考えられる再発性視神経炎や再発性脊髄炎)症例における、難治性の吃逆・嘔気の臨床的意義について検討を行った。対象とした35例中15例(43%)に、難治性の吃逆・嘔気の既往があった。その15例での35回の再発エピソードのうち、吃逆は66%、嘔気は80%の再発で確認された。吃逆・嘔気は、主要な神経症状よりも前駆して生じることが多く(54%)、さらに、吃逆・嘔気に前駆して、しばしば感冒様症状が出現していた。また、感冒様症状に引き続き難治性吃逆を生じた1例で、抗AQP4抗体価の推移を確認したところ、吃逆が生じた時点で著明な抗体価の上昇を認めていた。 最近の知見は、抗AQP4抗体がNMOの病態に深く関わることを、強く示唆している。特に、NMO病変の好発部位はAQP4の高発現部位と一致しており、その病変部位ではAQP4分子の消失が認められる(Misu et al. Brain 130:1224-34, 2007)。吃逆・嘔気に関わる延髄背側部(最後野Area postrema:AP)もAQP4の高発現部位であり、かつ、解剖学的な血液脳関門を欠いている(図)。このため、抗体の影響をより受けやすい可能性があり、それが神経症状に前駆する吃逆・嘔気に結び付いているのではないか、と推測された。すなわち、ウイルス感染→抗体価の上昇→吃逆・嘔気の出現→神経症状の出現、という仮説が考えられ、吃逆・嘔気の段階での治療介入により、引き続く失明や四肢麻痺を回避できる可能性も考えられた。
ALSラットモデル脊髄におけるコンドロイチン硫酸プロテオグリカン沈着
"Accumulation of chondroitin sulfate proteoglycans in the microenvironment of spinal motor neurons in amyotrophic lateral sclerosis transgenic rats"
○水野秀紀 (Mizuno H, Warita H, Aoki M, and Itoyama Y. J Neurosci Res, Published Online: 25 Apr 2008 )
筋萎縮性側索硬化症(ALS)は運動ニューロンの選択的な細胞死が引き起こされ、全身の萎縮と脱力が進行する原因不明の難治性神経変性疾患である。最近、運動ニューロンの変性にはアストロサイトなどのグリア細胞が大きく関与することが注目され、運動ニューロンをとりまく細胞外微小環境が注目されている。また、近年までに成体中枢神経系に神経幹/前駆細胞が存在することが明らかとなり、ALSに対してもそれらを用いた神経再生療法が新たな治療戦略として関心を集めている。
このような中で我々は、細胞外基質の主要構成成分として知られ、神経軸索再生阻害活性や細胞遊走阻害活性をもつコンドロイチン硫酸プロテオグリカン(CSPG)に注目、中枢神経系に発現するneurocan, versican, phosphacanのALSでの病態進行に伴う変化を解析した。我々の教室で開発されたALSモデルラット(His46Arg変異SOD1遺伝子導入ラット; Nagai, M, et al. J Neurosci 2001)を用い、免疫組織化学的手法で解析したところ、主病変部位である前角部を中心に有意なCSPG沈着亢進が明らかとなった。(図1) なかでもneurocanは発症前から始まる進行性の沈着を示し、発症後期で成体脊髄では生理的にほとんど検出されない全長型のisoformを検出した。 またCSPGとアストロサイトとの共局在が認められ、同細胞のCSPG沈着への関与が示唆された。(図2)
本研究によって、ALSのような慢性進行性の変性病態でのCSPG沈着が明らかとなり、反応性アストロサイトとともに再生に対して非許容的な環境を形成している可能性が示唆された。将来的な再生療法開発には、このような細胞外微小環境を再生しやすい場へと変化させる戦略も考えられ、今後CSPG沈着亢進機構の解明やCSPG分解をもたらす介入研究が必要と考えられた。
ジストロフィン複合体から局在移行したnNOSが産生するNOにより尾部懸垂での筋萎縮が引き起こされる
社会の高齢化に伴い、寝たきりや脳卒中罹患後の筋萎縮への対策が重要度を増している。近年、筋萎縮においてユビキチン・プロテアソーム系を介した蛋白分解経路の活性化が起こることが明らかになったが、筋萎縮の分子機構の詳細、特に筋細胞膜の機械的安定性の維持や細胞内情報伝達に非常に重要な分子であるジストロフィンを中心とした蛋白複合体の筋萎縮への関与は明らかではなかった。我々は不動による筋萎縮モデルの一つである尾部懸垂モデルを用いて、筋萎縮の分子機構を検討した。
筋萎縮の過程でジストロフィン複合体の一員として筋細胞膜に局在し細胞内情報伝達に重要な役割を担う神経型一酸化窒素合成酵素(nNOS)が、細胞質に局在を移すことを見出した(図1)。さらに電子常磁性共鳴測定器(EPR spectrometer)を用いて、萎縮筋における一酸化窒素(NO)合成量が増加していることをex vivoで直接示した。NOは転写因子Foxo3aの活性化を介してユビキチンligaseであるMuRF-1やatrogin-1の発現増加を引き起こしていた。nNOS阻害剤によって筋萎縮は軽減した。(図2)これらの結果からnNOS由来のNOが尾部懸垂モデルにおける筋萎縮促進分子として働いていることが明らかとなった(図3)。
nNOS/NOやそれらと相互作用する分子は筋萎縮対策の新たなターゲットの候補となる可能性がある。今後はALSなどの筋萎縮をきたす神経変性疾患で同様の機構が働いているかを解析し、治療応用が可能かどうか検討していく。