

研究内容
東北大学脳神経内科における研究
わたしたちは「新しいことに取り組もう」というかけ声のもと、東北大学神経内科としてのミッションを果たすため、日々研究に取り組んでいます。そして、今日も多くの大学院生が独立した研究者をめざし、教員に学んでいます。
教室の研究活動は、広く地域の先生方から託された患者さんの診療に端を発しています。また、公的研究費、素晴らしい共同研究者の方々・患者さん達のご協力と励ましによって支えられています。そのような心強い支援のもと、病因遺伝子を同定、iPS細胞を樹立、病態モデルを自らの手で確立し、新たな診断・治療法を開発するという一連の研究を展開できる私たちはとても恵まれているといえます。
感謝の気持ちを熱意にかえ、今後も代々引きついできた「研究魂」を教室員一丸となって発揮し、橋渡し研究(TR)の実践という形で社会に還元してまいります。まだまだ若手医師の皆さんの力が必要です。どうぞ、まずは一度見学にいらしてください。
医局長 ・ 菊池昭夫
■ 当教室の研究業績はPubMedをご覧下さい。
パーキンソン病関連疾患研究チーム(PDチーム)
長谷川 隆文*、菊池 昭夫、菅野 直人、吉田 隼(総合南東北病院)、小林 潤平(NHO米沢病院)、江面 道典、石山駿、伊藤友江(技能補佐員)(*チームリーダー)
大学院入学を希望される皆さんへ

わたしたちの研究チームは分子生物学、細胞生物学から機能画像解析、神経心理学といった多角的な手法を駆使し、パーキンソン病(PD)および関連疾患の病態解明と早期診断・進行抑制治療の確立を目指しています。大学院では研究技術の指導はもちろんのこと、分析力、批判的思考力、問題解決力および科学的表現力を養い、世界に通用する自立した研究者となるために必要な基礎的能力を育成します。チャレンジ精神溢れる意欲的人材を歓迎します。
- 課題1. メンブレントラフィック障害に着目したパーキンソン病の分子病態解析
-
研究内容1. 現代社会において、原材料調達から生産・販売に至るまでの物の流れを一元管理して、全体を最適化させるロジスティックスは必要不可欠な物流インフラとなっています。同様に、生体を構成する細胞には高度に専業化された物流システムであるメンブレントラフィック系が存在し、オルガネラ間あるいは細胞内外で積荷分子の輸送を繰り返し生命活動を下支えしています。受容体発現調節、シナプス伝達、タンパク分泌、オートファジー分解などメンブレントラフィックの役割は枚挙に遑がありません。近年、パーキンソン病(PD)やアルツハイマー病、筋萎縮性側索硬化症をはじめとする神経変性疾患の原因遺伝子の多くがメンブレントラフィック制御因子であることが相次いで判明し、病態解明と新たな治療法確立への糸口として注目を集めています(図1)。我々は初代培養神経細胞やショウジョウバエ・コンディショナルノックアウトマウスなどを駆使し、エンドソーム関連遺伝子VPS35、DNAJC13のミスセンス変異による家族性PD(PARK17, PARK21;文献2,3,5,9)や、家族性ALSおよび痙性対麻痺の原因となるエンドソーム輸送制御因子ESCRT(endosomal sorting complex required for transport)複合体(文献8)の機能異常が、αシヌクレイン(αS)、アミロイドβやTDP-43といった変性疾患関連タンパクの細胞内輸送・リソソーム・オートファジー分解に重要な役割をもつことを明らかにしてきました。上記遺伝子の一部(VPS35、LRRK2、Rab7L1)は互いに機能的相関を示すことが示唆されており、PD発症に至るcommon pathwayとなっている可能性があります。現在さらなる病態メカニズム解析を続けています。 図1. 家族性PD原因遺伝子(赤斜体文字)の多くはメンブレントラフィック制御分子である(文献4より引用)。 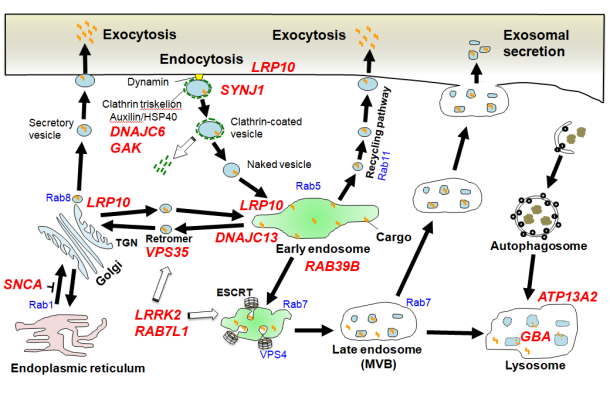
- 課題2. 異常タンパク細胞間伝播(プリオン仮説)の分子機構解明と進行抑制治療法開発
-
研究内容2. PD、レビー小体型認知症(DLB)、多系統萎縮症(MSA)は、神経変性部位に一致して異常凝集したαSタンパクが蓄積することから、シヌクレイノパチーと総称されています。今から十数年前、PD患者を対象として胎児中脳由来ドパミン神経細胞の移植手術が実施された時期がありました。残念ながらこの治療は内服によるドパミン補充療法に勝る有用性が確認されず、その後実施されることが無くなりました。近年になり、この移植治療を受けた患者脳を病理学的に詳細に検討した結果、ドナーである胎児由来の神経細胞内にαS陽性の凝集体が確認されたという驚くべき事実が報告されました。この発見を端緒として、凝集化αSが神経あるいはグリア細胞間を「飛び火」することで脳内病変が拡大するのではないか?という新たな病態仮説(プリオン仮説)が提唱される様になり一躍注目を集めました。その後この現象はアルツハイマー病(AD)、ALSなど異常タンパク凝集を共通病理とする他の神経変性疾患にも認められるとの実験的証拠が示され、変性疾患の共通病態として認識されつつあります。我々は早くからこの仮説の重要性に着目し、細胞間伝播現象の背景にあるαS吸収、分泌および分解のメカニズムを世界に先駆けて明らかにして来ました(図2A、文献5,10)。この過程において、凝集化αSの細胞内吸収は、ダイナミン依存性のエンドサイトーシスにより制御されることを見いだすと共に、同過程を阻害する抗うつ薬の一種セルトラリン投与により、神経・グリア細胞内へのαS吸収が有意に抑えられることを発見しました。現在、αS脳内伝播を再現したマウスモデルを用い、治療薬としての効果検証を進めています(図2B)。これと並行して、神経・グリア細胞表面に発現する線維化αS受容体の網羅的解析にも取り組んでいます。将来的には線維化αS受容体に対する特異抗体や結合阻害分子を用いたより選択的・特異的なシヌクレイノパチー進行抑制治療の開発を目指したいと考えています。 図2. A, 神経・グリア細胞におけるαS吸収・分解・分泌の制御メカニズム(文献5より一部改変引用)。B, 線維化αS脳内伝播マウスモデルを用いた進行抑制治療法の開発。 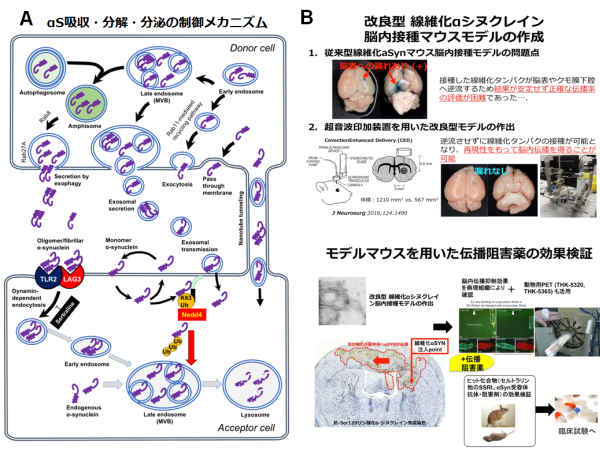
- 課題3. 新規PETプローブを用いた各種神経変性疾患の画像診断法確立
-
研究内容3. 神経変性疾患では、病変組織の細胞内外に各々の疾患を特徴付ける異常タンパク質の凝集・蓄積が観察され、症状進行に伴ってその範囲が拡大してゆくことが知られています。具体的には、PD・DLB・MSAではαS、アルツハイマー病(AD)・大脳皮質基底核症候群(CBS)・進行性核上性麻痺(PSP)においてはタウ凝集体の脳内沈着がみられます。生体脳においてこれらの異常凝集タンパクを可視化する手法として、近年ポジトロン断層法(positron emission tomography:PET)検査の有用性が注目されています。我々は、BF研究所および本学で開発された[11C]BF227 や[18F]THK5351 PETプローブを用い、MSA患者脳のαS凝集体 (図3A) やCBS患者脳におけるタウ凝集体・モノアミン酸化酵素B (図3B) の画像化を世界に先駆けて成功して来ました(文献6)。今後より感度・特異度に優れた新規PETプローブを用い、各種神経変性疾患における脳内異常タンパク病変の可視化に取り組む予定です。これらの取り組みを通じて、早期診断や病期の把握、疾患修飾療法の治療効果判定といった臨床応用を目指しています。 図3. A, MSA患者の[11C]BF227 PET画像。健常者(上段)と比較して、MSA患者(下段)では大脳白質・基底核などの領域で[11C]BF227 PETトレーサーの集積が観察される。B, CBS患者の[18F]THK5351 PET画像 (文献5より引用)。健常者(左)と比較しCBS患者群(右、症例1?3)では片側の大脳皮質・基底核の一部領域において[18F]THK5351 PETトレーサーの集積増加(赤い部分)を認め、臨床症状の左右差とも一致していた。(文献6より一部改変引用) 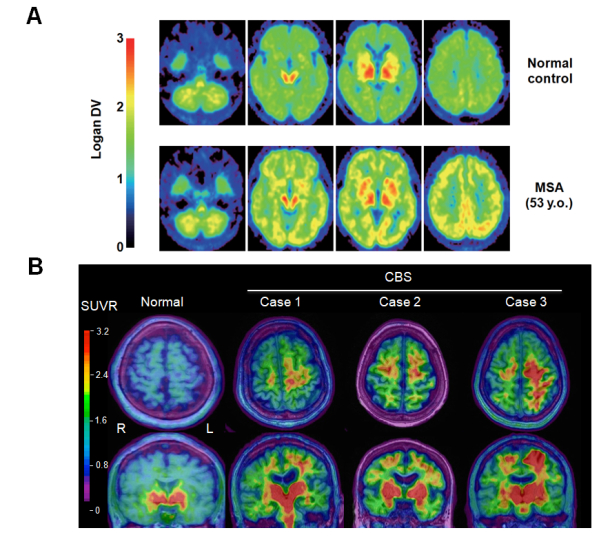
- 課題4. 核内αSの機能探索:αSを介するヒストン修飾機構の解明
-
研究内容4. αSはPD発症に関わる分子病態において中核となるタンパクであり、αSの神経毒性により細胞死が生じると考えられています。我々はこれまでにαSがどのように細胞内で運ばれるか(課題1)、どのように細胞間を移動するか(課題2)、それを実際の患者さんでどのように評価するのか(課題3)研究してきました。ではそうして細胞内外を移動したαSはどのように細胞毒性を発揮するのでしょうか? PDの神経細胞にαSが蓄積する事が報告されてから今日に至るまでのおよそ20年間、多くの研究がなされてきましたがいまだ決定的な機構は不明のままです。解明のためには新たなアプローチが必要かもしれません。そもそもsynucleinの名前は、ゴマフシビレエイ発電器官の神経前終末と核に局在するタンパクとして同定されたことに由来します (synapse+nucleus=>synuclein)。これまでシナプスにおける役割はある程度解明されつつあるものの、核内αSの生理的および病的意義はいまだ十分にわかっておりません。細胞における核の役割は様々ですが、とりわけ重要なのは転写調整を通したタンパク発現制御です。タンパク発現制御は神経細胞の恒常性維持に重要であり、転写の異常が神経変性をもたらすことは容易に想像できるかと思われます。近年、ヒストン修飾やDNAのメチル化といった“エピジェネティクス”による転写制御機構が脚光を浴びており、我々はαSがエピジェネティクスに与える影響を検討しています。ここまでで、αSがヒストンH3の9位リシンのメチル化(H3K9me2)を増強する事を確認しました(文献7)。H3K9me2はDNAの高次構造を密にするため転写因子が該当DNAに結合することができなくなります(図4A)。さらに、クロマチン免疫沈降(ChIP)と転写産物のスクリーニングによって、SNAP25とL1CAMという2つの遺伝子の転写がαSによって抑制される事を見いだしました(図4B)。特にSNAP25はシナプス制御において重要なタンパクであり、αSはエピジェネティクスを介して神経機能を抑制している可能性があります。αS関連の遺伝子は他にも多数あると推定され現在網羅的解明を進めています。 図表ファイル名:A. ヒストンH3タンパクの9位、27位のメチル化は転写抑制を意味します。met: メチル化、ac: アセチル化 B. αSはエピジェネティクスを介してSNAP25とL1CAMの転写を抑制します。 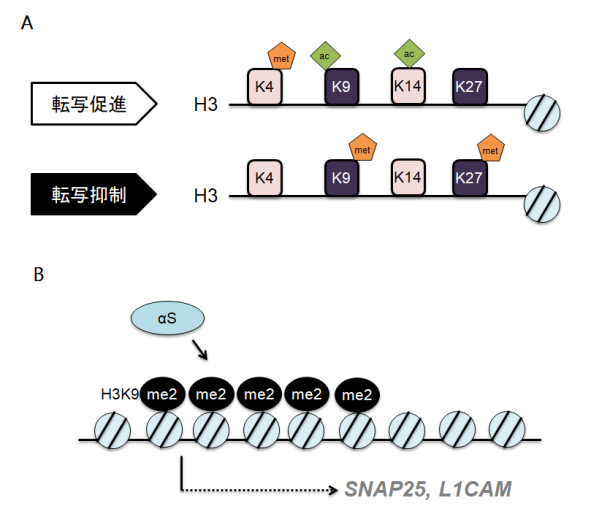
- 主な研究業績とリンク(PDチーム)
-
過去3年程度の主な研究業績 1. Kobayashi J, Hasegawa T, Sugeno N, Yoshida S, Akiyama T, Fujimori K, Hatakeyama H, Miki Y, Tomiyama A, Kawata Y, Fukuda M, Kawahata I, Yamakuni T, Ezura M, Kikuchi A, Baba T, Takeda A, Kanzaki M, Wakabayashi K, Okano H, Aoki M. Extracellular α-synuclein enters dopaminergic cells by modulating flotillin-1-assisted dopamine transporter endocytosis. FASEB J 2019 33: 10240-10256. 2. Ezura M, Kikuchi A, Ishiki A, Okamura N, Hasegawa T, Harada R, Watanuki S, Funaki Y, Hiraoka K, Baba T, Sugeno N, Oshima R, Yoshida S, Kobayashi J, Kobayashi M, Tano O, Nakashima I, Mugikura S, Iwata R, Taki Y, Furukawa K, Arai H, Furumoto S, Tashiro M, Yanai K, Kudo Y, Takeda A, Aoki M. Longitudinal changes in 18F-THK5351 PET in corticobasal syndrome. Eur J Neurol 2019 26:1205-1211. 3. Carballo-Carbajal I, Lagun A, …, Hasegawa T., et al., Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat Commun 2019; 10: 973. 4. Yoshida S., Hasegawa T., Suzuki M., et al. Parkinson’s disease-linked DNAJC13 mutation aggravates α-synuclein-induced neurotoxicity through perturbation of endosomal trafficking. Hum Mol Genet. 2018; 27, 823-836. 5. Hasegawa T., Yoshida S., Sugeno N., et al. DnaJ/Hsp40 family and Parkinson’s disease. Front Neurosci. 2018; 11, 743. 6. Willen K., Edgar J.R., Hasegawa T., et al. Aβ accumulation causes MVB enlargement and is modelled by dominant negative VPS4A. Mol Neurodeg. 2017; 12:61. 7. Hasegawa T., Sugeno N., Kikuchi A., et al., Membrane trafficking illuminates a path to Parkinson’s disease. Tohoku J Exp Med. 2017; 242, 63-76. (invited review) 8. Kikuchi A., Okamura N., Hasegawa T., et al. In vivo visualization of tau deposits in corticobasal syndrome by 18F-THK5351 PET. Neurology. 2016; 87, 2309-16. 9. Sugeno N., Jackel S., Voigt A., et al., α-Synuclein enhances histone H3 lysine-9 dimethylation and H3K9me2-dependent transcriptional responses. Sci Rep. 2016; 6, 36328. 10. Oshima R., Hasegawa T., Tamai K., et al. ESCRT-0 dysfunction compromises autophagic degradation of protein aggregates and facilitates ER stress-mediated neurodegeneration via apoptotic and necroptotic pathways. Sci Rep. 2016; 6, 24997. - 大学院入学を希望される方へ
-
神経内科疾患には頭痛やてんかん、脳卒中といったcommon diseaseが多い一方、未だ病態不明で根治療法の確立していない難病も多数存在します。みなさんの中にも、「いつの日か難病を克服してみせる」という信念をもって医師の道を志された方がおられることでしょう。また、診療現場で日々直面する難題に対して、新たな診断法や治療・予防法を考案したいという衝動をもたれた方もいらっしゃるかと思います。一昔前まで、神経内科疾患の多くは原因不明であり、病気のメカニズムも難解で攻略の糸口を見出しがたいものでした。しかし、近年の分子遺伝学、細胞生物学、脳機能画像などの研究進歩により事態は一変し、多くの神経難病の病態が今まさに分子レベルで解明されつつあり、大変魅力的な研究対象となっています。また、診断・治療法の進歩に伴い、科学的思考と基礎医学的素養は、臨床現場でも要求される様になっています。研究医を志す諸君は勿論のこと、将来臨床医として活躍したいと考えておられる方にも、是非大学院で一定の期間研究に従事し、論理的思考力を培うことを推奨したいと思います。
PDチームのメンバーは皆明るく気さくなfunky guyで、熱いresearch mindをもっています。疑問を感じたり困ったときは、気兼ねなく経験豊富な先輩に相談できる風通しの良さが自慢です。諦めず最後まで研究をやり遂げ、論文化したときの達成感は何物にも代え難いです。地位や名誉、お金はあの世には持って行けませんが、血と汗と涙の結晶である論文は永遠に残り引用され、後生の人々に受け継がれます。人生は一度きり、この世に生きた証をしっかりと残そうじゃありませんか。さあ、一緒にリサーチしましょう!関連リンク(PDチーム)
https://www.researchgate.net/profile/Takafumi_Hasegawa
http://researchmap.jp/read0163728
http://lewybody2016.jp/member.html
http://www.med.tohoku.ac.jp/enc/med/awardees2010.html
https://www.tohoku.ac.jp/japanese/2018/01/press20180117-01.html
https://www.tohoku.ac.jp/japanese/2016/10/press20161031-02.html - 課題1. MOG抗体、AQP4抗体に関する臨床的研究
-
MOG抗体、AQP4抗体に関する臨床的研究 視神経脊髄炎(neuromyelitis optica: NMO、別名Devic病)は、視神経と脊髄を病変の首座とする中枢性炎症疾患です。私たちは、主にNMOや多発性硬化症(MS)患者の血清中の自己抗体の検索を行っています。2004年Mayo clinicとの共同研究により、中枢神経系の軟膜下や血管周囲に特異的に反応する抗体NMO-IgGが見いだされ、その後対応抗原がアクアポリン4(AQP4)であることが判明しました。本邦における多くの炎症性脱髄性疾患の診断に大きく貢献し、NMOやMSの血清や髄液中の自己抗体を判定し、臨床的特徴や治療法について研究しています。
私たちは、従来の脱髄疾患の枠を超えたアストロサイトパチーという疾患概念を提唱し、その臨床的特徴や病態、治療法などを提唱してきました(1, 2, 3)。ミエリンオリゴデンドロサイト糖蛋白(MOG)抗体は古くから脱髄との関連が指摘されてきましたが否定的な論文も多くその意義は不明でしたが、近年細胞外ドメインを認識する抗体の特異度が高いことから報告が相次いでいます。当科で行った検討で、MOG抗体は急性散在性脱髄性脳脊髄炎(ADEM)の他、AQP4抗体を有しない長い脊髄炎や両側視神経炎を呈するNMO関連疾患(NMOSD)でも陽性例があることを初めて報告しました(4, 5)。
脱髄性疾患は、本来中枢神経系において髄鞘を密に構成する白質を主体に生じる疾患と考えられてきましたが、私たちのMOG抗体陽性例の検討によって、特にてんかん発作を伴い皮質表層の片側性ないし両半球内側性皮質などに特徴的な病変を呈する一群が存在することが近年明らかとなりました(6, 7)。脱髄疾患の概念自体に大きな影響を与えうる事実であり、本研究はアメリカ神経学会雑誌の2017年3月号の巻頭言および表紙を飾りました(6)(図1)。なぜ抗MOG抗体が、このような皮質病変を起こすのかは不明であり、今後も病態など多角的に研究を進めています。図1. 抗MOG抗体における皮質性病変。脳溝に沿った信号変化と血流増加を特徴とする。 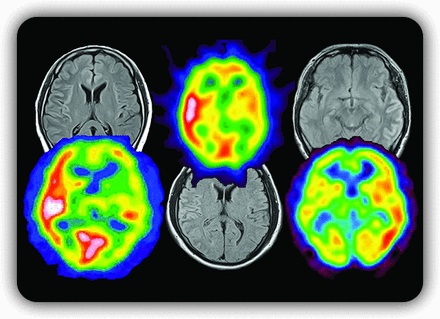
- 課題2. 炎症性脱髄性疾患の病態に基づいた病勢や予後に関するバイオマーカーの研究
-
炎症性脱髄性疾患の病態に基づいた病勢や予後に関するバイオマーカーの研究 NMO関連疾患(NMOSD)の再発急性期では、アストロサイトマーカーであるGFAPが脳脊髄液中で著明に上昇し、MSなど他疾患との鑑別に有用である他、再発後の予後予測因子にもなり得ることを報告しています(図2: NMO再発急性期の脳脊髄液中GFAPの著明上昇)。NMOSDではAQP4抗体がアストロサイト足突起を補体介在性・補体非介在性の2つの経路で傷害していくことが分かっています(8)。今後はより簡便かつ鋭敏にアストロサイト傷害を反映するバイオマーカーの検索を進めています。近年、MOG抗体による様々な神経症状を起こす疾患(MOG抗体関連疾患)がトピックとなっています。MOG抗体陽性疾患ではNMOSDと異なり、脳脊髄液中GFAPを認めません(図3: MOG抗体陽性例、AQP4抗体陽性例、MS、コントロールでの脳脊髄液中GFAP値)。MOG抗体関連疾患がNMOとは異なる病態であることが明確に解ってきています(9)。
多発性硬化症(MS)は主に若年女性に発症する自己免疫性疾患で、本邦にも現在約2万人の患者が罹患していると言われています。NMOと比較して多くの研究が長年行われていますが、未だに原因が殆ど解っていません。初めに世界で認められたインターフェロン療法やコパクソン、近年保健収載されたジメチルフマル酸も、有効性は明らかですがその作用機序には諸説あります。MSにおいては病態本質の不思議さもあって、パイロット的な治験薬が数々登場しては消えています。私たちは、ジメチルフマル酸が酸化的ストレスに関わるNrf2系に作用することに注目し、現在治療前後における様々な炎症・酸化ストレス関連の疾患活動性バイオマーカーの検索を行っています。図2. NMOにおけるアストロサイトパチー 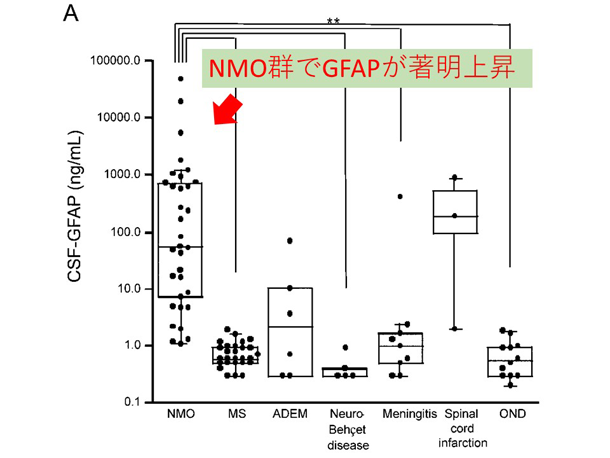
図3. MOG抗体陽性群はGFAP上昇しない 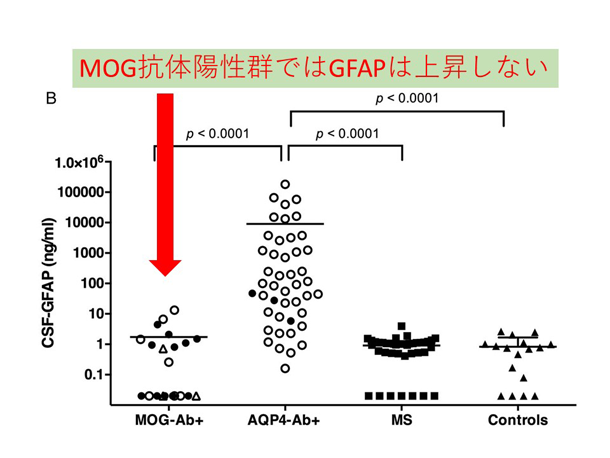
- 課題3. 中枢性炎症性脱髄疾患における病理学的研究−アストロサイト障害に伴う脱髄機序の解明
-
中枢性炎症性脱髄疾患における病理学的研究−アストロサイト障害に伴う脱髄機序の解明 わたしたちの研究チームは脱髄疾患の病理学的研究を進め、NMOやMS、Balo病の病理学的特徴を明らかにしてきました(10−13)。NMO は、抗アクアポリン4(AQP4)抗体と呼ばれる自己抗体により生じる中枢神経の炎症性疾患です。AQP4はアストロサイトに発現しているため、抗AQP4抗体によってアストロサイトが破壊されるという病態がNMOの本質と考えられています。しかし、麻痺などの臨床的障害は、その結果生じる髄鞘や神経軸索の脱落及び変性が原因と考えられますが、その規序はまだ不明です。
私たちはアストロサイトパチーに伴って生じる脱髄規序を解明するため、人の病理検体と動物モデルを用いて検討を行っています。NMOの動物モデルは、他施設との共同研究によって独自に開発した、マウスに対し高親和性を示す抗AQP4抗体を用いて作成します。この抗体をマウスの脳に直接注入する、あるいは炎症を起こしたマウス・ラットの腹腔内に注入することで、NMOと類似の病変を作成することが可能であり(図4,5)、現在その病態解析を行っています。
アストロサイト障害によって、二次的な脱髄がその中心部に生じることや、炎症細胞の浸潤と脱髄の範囲が相関すること、脱髄範囲の分布が神経の走行に沿って拡大していくことなど、ユニークな現象が発見されています(未発表)。現在は、さらに脱髄の根本的な分子メカニズムを解明するために研究を続けています。図4. 血管周囲アストロサイトパチー(AQP4染色) 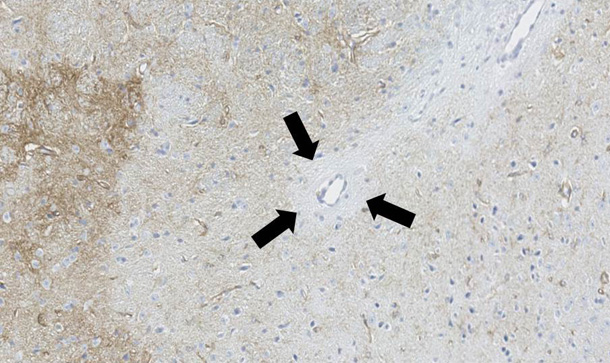
図5. 病変の経時的変化(縦軸:病変面積,横軸:注入後経過 (日数) ) 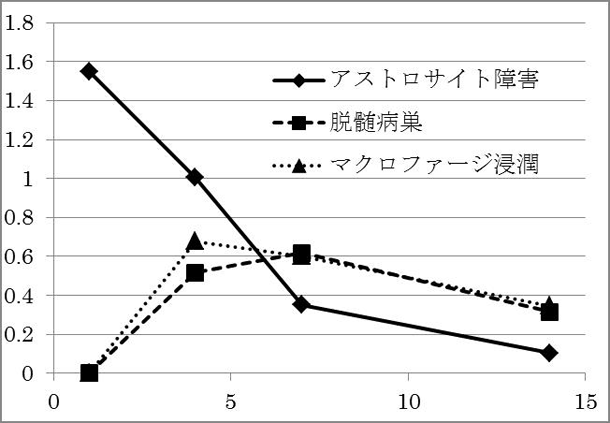
- 主な研究業績とリンク(MSチーム)
-
1. Takai Y, Misu T, Suzuki H, et al. Staging of astrocytopathy and complement activation in neuromyelitis optica spectrum disorders. Brain. 2021; in press. 2. Takai Y, Kuroda H, Misu T, et al. Optimal management of neuromyelitis optica spectrum disorder with aquaporin-4 antibody by oral prednisolone maintenance therapy. Mult Scler Relat Disord. 2021 Jan 22;49:102750. 3. Namatame C, Misu T, Takai Y, et al. CH50 as a putative biomarker of eculizumab treatment in neuromyelitis optica spectrum disorder. Heliyon. 2021 Jan 8;7(1):e05899. 4. Matsumoto Y, Misu T, Mugikura S, et al. Distinctive lesions of brain MRI between MOG-antibody-associated and AQP4-antibody-associated diseases. JNNP. 2020 Dec 30:jnnp-2020-324818. 5. Takai Y, Misu T, Kaneko K, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study. Brain. 2020 May 1;143(5):1431-1446. 6. Hillebrand S, Schanda K, Nigritinou M, Tsymala I, Bohm D, Peschl P, Takai Y, Fujihara K, Nakashima I, Misu T, Reindl M, Lassmann H, Bradl M. Circulating AQP4-specific auto-antibodies alone can induce neuromyelitis optica spectrum disorder in the rat. Acta Neuropathol. 2019 Mar;137(3):467-485. 7. Kaneko K, Sato DK, Nakashima I, et al. CSF cytokine profile in MOG-IgG+ neurological disease is similar to AQP4-IgG+ NMOSD but distinct from MS: a cross-sectional study and potential therapeutic implications. JNNP. 2018 Sep;89(9):927-936. 8. Nishiyama S, Misu T, Shishido-Hara Y, et al. Fingolimod-associated PML with mild IRIS in MS: A clinicopathologic study. Neurol Neuroimmunol Neuroinflamm. 2017 Nov 10;5(1):e415. 9. Ogawa R, Nakashima I, Takahashi T, et al. MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neuro Neuroimmunol Neuroinflam 2017; e322. 10. Fujimori J, Takai Y, Sato DK, et al. Bilateral frontal cortex encephalitis and paraparesis in a patient with anti-MOG antibodies. JNNP. 2017; 88: 534-536. 関連リンク(MSチーム)
東北大学多発性硬化症治療学講座
- 課題1. ALSを対象とした橋渡し研究(translational research)の実践
-
ALSを対象とした橋渡し研究(TR)の実践 ALSは、おもに運動神経の変性によって全身の筋肉にやせと筋力低下が進行し、やがては呼吸筋麻痺にいたる難治性の神経疾患です。
ALSの進行を抑制する薬剤としてリルゾールとエダラボンがありますが、症状を改善する治療法はまだありません。このため、新しい治療法の開発が世界中で切望されています。私たちは、日本で発見された神経栄養因子である肝細胞増殖因子(hepatocyte growth factor, HGF)を用いたALS治療法の開発にとり組んでいます。
2011年、世界初となるHGFの脊髄腔内投与による第I相試験を東北大学病院で開始し、2014年までに安全性と薬物動態を確認しました。この成果をもとに現在、ALSに対するHGFの有効性と安全性を確認するための第II相試験(医師主導治験)を実施しています。
このHGF治験(第II相)では、東北大学病院、大阪大学医学部附属病院で合計48例のALS患者さんにご参加いただく計画で、ヒト遺伝子組換えHGFタンパク質製剤(クリングルファーマ社より提供)を使用します。2週間に1回、外来通院しながら脊髄腔内へくり返し治験薬を投与するため、皮膚の下に埋め込むポートと専用のカテーテルを脊髄腔にうめこむ手術が必要です(治験終了時にはとりのぞく手術も必要です)。有効性をきちんと評価するため、最初の6か月間はご本人にも治験担当者にも実薬(HGF)か偽薬(プラセボ)かを知らされない「二重盲検」という方法で実施します。
この治験は2019年8月までを予定しています。ご参加いただくのは本治験計画で定められた基準(条件)に合うALS患者さんに限られるため、神経内科専門医の先生方に全国からご協力いただきながら現在進行中です。
お問い合わせ先など、詳しくは下記リンクをご覧ください。 (割田 仁,井泉瑠美子,池田謙輔,鈴木直輝)
東北大学「プレスリリース」
TOPページより「ニュース」該当記事
UMIN-CTR
- 課題2. GNEミオパチーに対するアセノイラミン酸の臨床試験(医師主導治験)
-
GNEミオパチーに対するNアセチルノイラミン酸の臨床試験(医師主導治験) GNEミオパチーは、体幹から離れた部位から筋肉が萎縮・変性する難治性の筋疾患です。病理学的に特徴的な縁取り空胞の存在が、別名である「縁取り空胞を伴う遠位型ミオパチー」という名前の由来となっています。
2001年に常染色体劣性遺伝によるGNE遺伝子の変異が病気の原因であることがわかりました。骨格筋の機能維持に重要なシアル酸の生合成低下が病気の本態でした。疾患モデルマウスが作成されシアル酸の一つであるNアセチルノイラミン酸(アセノイラミン酸)の経口補充がミオパチー症状の改善に有効であることが明らかとなりました。
前臨床試験の結果を元に2010年から東北大学病院において、世界に先駆けてシアル酸製剤(アセノイラミン酸)の薬物動態と安全性を評価する第I相試験を行いました。薬効を評価するための第II/III相の臨床試験を日本医療研究開発機構(AMED)の支援の下、2016年3月より開始しています。
当科を含む国内5施設で計20名のGNEミオパチー患者さんにご協力いただき、48週間の投与期間でのプラセボを対照とした二重盲検試験を行い、現在も延長試験が進行中です。当科では厚生労働省の難治性疾患政策研究事業「希少難治性筋疾患に関する調査研究班」も運営しています。オーファンドラッグの実用化に向けて臨床調査・応用研究の両面を推進していきたいと思います。 (鈴木直輝)
- 課題3. 家族性ALSの遺伝学的背景の解明
-
家族性ALSの遺伝学的背景の解明 筋萎縮性側索硬化症(ALS)は運動ニューロンの選択的な細胞死をひき起こす成人発症の神経変性疾患です。患者の約10%は家族性に発症がみられます(家族性ALS)。この家族性ALSの原因遺伝子は1993年にSOD1遺伝子が同定されて以降、近年の次世代シークエンサー注1の開発に伴い、25種類以上の原因遺伝子が報告されています。
東北大学神経内科では1991年以来111家系の日本人家族性ALSを集め、その原因遺伝子を探索してきました。直接塩基配列決定法(サンガーシークエンス注2)を用いて36家系にSOD1変異、12家系にFUS変異を同定してきました。さらに、最近、遺伝医療学分野との共同研究によりALS関連35遺伝子を標的とした網羅的解析を加え、全体の約半数の家系に原因遺伝子変異を明らかにしました(図1,3)。
残る半数の自験家系では未だ原因遺伝子が解明されておらず、最近の解析で見出された病原性が未確定の候補遺伝子変異が病的意義をもつ可能性と、未知のALS原因遺伝子変異が存在する可能性が示唆されています。本研究では、網羅的遺伝子解析研究の推進と、新規候補遺伝子変異の病原性を明らかにすることを目的としています。
原因遺伝子未同定の家系を対象に、次世代シークエンサーを用いた全エクソン解析による新たな候補遺伝子変異の探索を行います。また、候補遺伝子変異を導入した形質の解析、健常ヒト人工多能性幹細胞(iPS)細胞由来運動ニューロンに変異を導入し、病態解明を目指しています。
これまでの解析で欧米人家族性ALS家系の30%近くに見出されるC9ORF72の反復異常伸長が自験家系には存在しないといった、欧米人とは異なる日本人ALSの遺伝学的背景を明らかにしており、ALSにおける分子病態の多様性が示唆されています。このような現状の元、原因遺伝子未解明の日本人家族性ALS家系の網羅的遺伝子解析により、新たな原因遺伝子を発見することでALS発症メカニズムを解明する手がかりを見出し、治療法の開発につながる病態研究を発展させることが期待されます。 (西山亜由美)
[注1] 次世代シークエンサー:数千万〜数億の DNA 断片の塩基配列を同時並列的に決定することで、短期間でギガ(10 億)単位での塩基配列を決定出来るシークエンサー。
[注2] サンガーシークエンス:酵素を用いて末端が特定の塩基に対応する DNA 断片を合成し DNA を構成する塩基配列を決定すること。
図1. 東北大学神経内科で集積した家族性ALS 111家系の原因遺伝子頻度.SOD1, FUS, SETX, TARDBP, ANG, OPTN, ALS2はそれぞれ変異が見出された家族性ALS原因遺伝子(本文参照).
”VUS”(variants of unknown significance)は、病的意義がまだ確定できないものの発症に関与する可能性がある変異.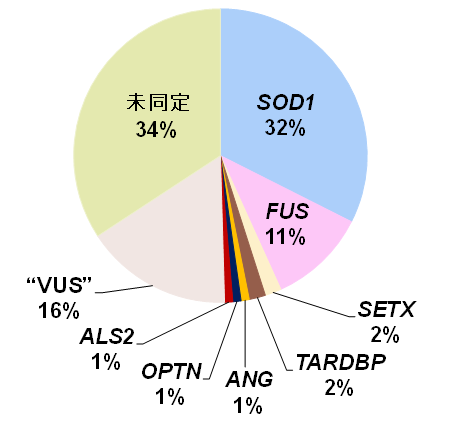
- 課題4. iPS細胞を用いたALSの軸索における病態の解明
-
iPS細胞を用いたALSの軸索における病態の解明 私たちは日本人の家族性ALSの中でFUS変異が2番目に高い頻度で認められることを報告しています(3, 7)。また孤発性ALSの特徴的な病理像として、脊髄前角運動ニューロン内のTDP-43蛋白質沈着が知られています。ALSの有力な病態仮説としてRNA代謝異常があり、FUSおよびTDP-43はいずれもRNA結合蛋白質です。私達はFUS変異およびTDP-43をコードするTARDBP変異に注目しました。
運動ニューロンは生体内では脊髄から手足まで伸びる長い軸索を経由して骨格筋を支配します。RNA結合蛋白質(FUSやTDP-43)の変異により軸索のRNA輸送や局所蛋白質翻訳の異常が生じると仮定し、ALSの軸索における異常を明らかにしたいと考えました。
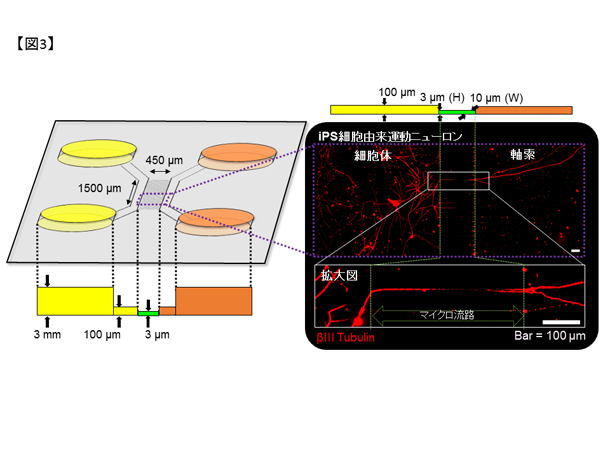
私達は慶應義塾大学との共同研究でFALS患者さん由来のiPS細胞注1を樹立すると共に、ゲノム編集技術を用いて変異以外が遺伝的に共通な細胞を作成しました(図2,6)。これらのiPS細胞を運動ニューロンへ分化誘導し、東京大学生産技術研究所との共同研究による広流路マイクロ流体デバイス注2を用いて軸索を分離・RNAシーケンス注3を行うことで軸索の病態に関連した異常を見出しています。さらに、運動ニューロンの形態異常や軸索輸送の障害を、狭流路マイクロ流体デバイス(図3)とmRNA標識蛍光分子(図4)を用いたライブセルイメージングで定量的に評価し、治療標的候補を見出しています。
今後は、新規マイクロ流体デバイスの特性を活かして蛋白質レベルの網羅的解析も予定しています。細胞・動物モデルでの介入実験を経て、治療薬開発につなげていきます。 (秋山徹也,光澤志緒,鈴木直輝)
[注1] induced pluripotent stem cellの略。人工多能性幹細胞とも言われ、目的細胞へ分化誘導可能な多能性を獲得した細胞です。体細胞に初期化因子(山中因子など)を発現させて作製されます。
[注2] ここでは、ニューロン観察用のデバイスを指します。両脇のチャンバーの間にあるマイクロ流路には細胞体は入ることができず、ニューロンの細胞体と軸索を分離して培養でき、軸索輸送などの研究に有用です。
[注3] RNA分子を網羅的に読み取る手法の一つです。
図2. iPS細胞とゲノム編集技術の組み合わせの概略。iPS細胞のiPS細胞によりヘテロ変異を持つ細胞(FALS)をコントロール細胞(健常者)と比較可能です。しかし、これらは同一人物由来ではないため、遺伝学的背景(年齢・性別・人種など)が異なり得ます。ゲノム編集技術によりコントロール細胞に変異を導入、及びFALS由来細胞の変異を修復し、FUSの変異の有無のみが異なる細胞を作成しました。
図3. マイクロ流体デバイス(#SND450-1)の模式図及びデバイス上で培養したiPS細胞由来運動ニューロンの染色像。運動ニューロンをマイクロ流路(緑の点線内)の左側へ培養すると、軸索のみが右側へ通過可能です。βIII tubulin: 神経細胞の代表的なマーカー。
図4. 蛍光分子の模式図。mRNAと結合しない場合は蛍光を吸収するクエンチャーと距離が近いため光らないが、ターゲットとなるmRNAと結合すると蛍光を呈します。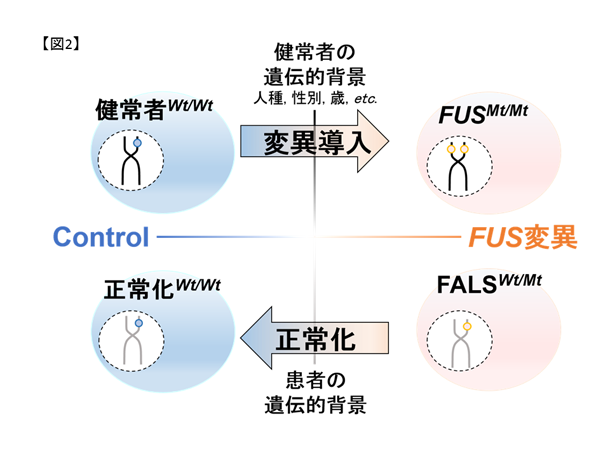
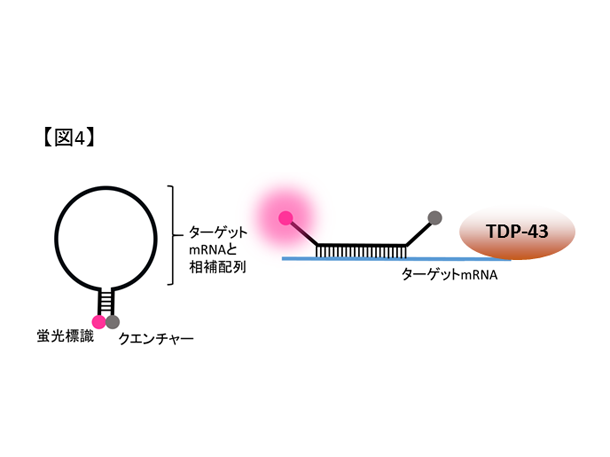
- 課題5. 細胞外微小環境に着目したALS神経再生療法の探索
-
細胞外微小環境に着目したALS神経再生療法の探索 ALSは、未だ根本的な治療法がない神経変性疾患のひとつです。臨床症状として、進行性・致死性の筋萎縮・筋力低下が生じます。脊髄では選択的な運動神経の変性・脱落と同時に、神経膠細胞(グリア細胞)の著しい増加が認められます。
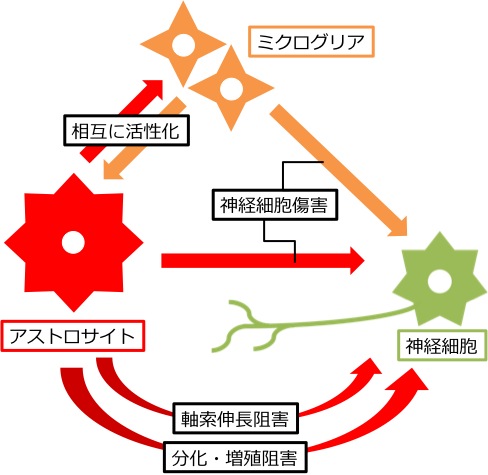
ALSにおける運動神経の変性が致死的となる原因のひとつに、成体哺乳類の神経細胞がそもそも再生しにくいうえ、成体脊髄では神経細胞を取り巻く細胞外の微小環境(グリア細胞を含む)が神経再生を阻害していることが挙げられます(下図,上段)。私たちは、この細胞外微小環境に着目し、生体にもともと内在している神経再性能を促進したり、分化増殖をコントロールしたりする治療戦略を探索しています。具体的には、主要な細胞外基質であり、神経再生阻害因子であるコンドロイチン硫酸プロテオグリカン(chondroitin sulfate proteoglycan: CSPG) や、神経幹細胞から神経細胞への分化を誘導もしくは阻害する分子群に着目しています。つまり、ALS病態の脊髄における細胞外微小環境を神経再生に寛容な状態へと変化させることにより、神経再生を促進したいと考えています。
日本人家族性ALSの原因遺伝子でもっとも頻度の高いSOD1の変異遺伝子を全身に過剰発現させたラットALSモデルは当教室で確立しました(Nagai M, et al. 2001)。現在までにALSモデルラットの脊髄で、CSPGの沈着が増えていること (Mizuno H, et al. 2008)、CSPG受容体が本来発現しないアストロサイトに発現していること(Shijo T, et al. 2018、下図, 下段,6)を発見しており、神経変性にともなう病的現象と考えています。今後、ALSモデルラットを用い、神経再生を促進し、ALS病態の進行を抑制する戦略を見出してゆくことを目標としています(下図,下段)。 (四條友望,割田 仁)

- 課題6. 多系統蛋白質症(MSP)の病態解明
-
MSPの病態解明 多系統蛋白質症(MSP;multisystem proteinopathy)は複数の異なる疾患(筋萎縮性側索硬化症や前頭側頭型認知症、封入体ミオパチー、骨パジェット病など)を組み合わせた形で発症する稀な優性遺伝性疾患です。これまでに、VCP、hnRNPA1など5つの疾患原因遺伝子が報告されています。これらの遺伝子は細胞質内でmRNAの翻訳制御に関わるRNA顆粒の生成/分解に寄与します。当教室は封入体ミオパチーの原因遺伝子としてhnRNPA1を同定しました(10)。
hnRNPA1遺伝子変異によりmRNAの翻訳制御が変化することが報告されていますが、複数の異なる臓器で選択的な細胞変性が生じる原因は明らかではありません。そこで、私達はMSP病態を再現するヒト細胞モデル・動物モデルを作製し,これらを用いた病態解明を目指しています。hnRNPA1変異MSP患者由来の疾患iPS細胞を樹立し、骨格筋細胞と神経細胞へとそれぞれ分化誘導しました。現在、それぞれの細胞を種々のストレス条件下で培養し、hnRNPA1陽性封入体の形成(図7)やRNA顆粒の生成/分解を中心に病態形成メカニズムの解明をめざしています。 (池田謙輔,割田 仁)
図7. 上段:患者由来骨格筋細胞の核周囲にataxin-2陽性顆粒を認める.下段:患者由来神経細胞の核周囲にhnRNPA1/G3BP陽性顆粒を認める.Ataxin-2とG3BPはともにRNA結合蛋白質. 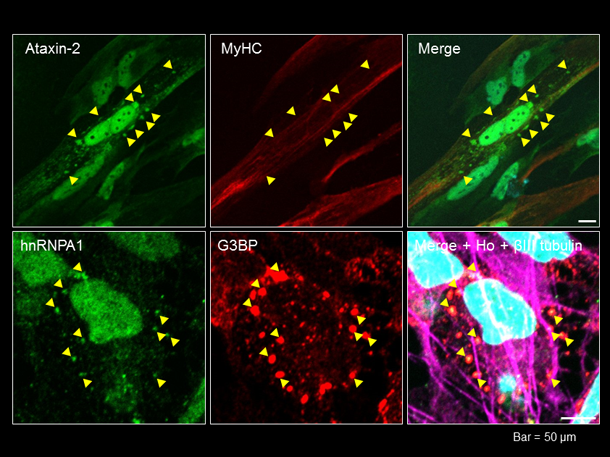
- 課題7. dysferlinopathy(dysferlin異常症)の遺伝学的背景の解明
-
dysferlinopathy(dysferlin異常症)の遺伝学的背景の解明 成人で発病する遺伝性ミオパチーのなかで患者数の多いものが、肢帯型筋ジストロフィーと遠位型ミオパチーです。その中でも、dysferlin遺伝子変異によっておこる肢帯型筋ジストロフィー2B型や三好型遠位型ミオパチーはdysferlinopathy(dysferlin異常症)と総称されています。dysferlin蛋白は、筋細胞膜の障害後の修復過程で重要な役割を持つ蛋白とされており、この機能が失われることで筋細胞の炎症や変性が生じると考えられています(図8)。
東北大学神経内科および研究協力施設の仙台西多賀病院では、1998年以降、海外や全国の研究施設・病院から、dysferlin異常症が疑われる患者の遺伝子検査の依頼を受け入れています。今まで、150人以上のPCR-SSCP法によるdysferlin遺伝子検査を行ってきましたが、約40%の症例では変異が確認されていませんでした。これらの症例では、二次的にdysferlin蛋白の発現低下を起こす別の遺伝子の変異が関連していることが予想されていましたが、従来の技術では、多数の遺伝子を網羅的に解析することが困難でした。
そこで、近年実用化され、網羅的遺伝子配列決定を可能とする次世代シークエンサーを用いることで、これらの未診断例における原因遺伝子を明らかとしたいと考えました。
dysferlin異常症が疑われながらも未診断である症例を対象として、筋疾患と関連することが知られている42遺伝子の翻訳領域を対象に遺伝子の配列決定(ターゲットリシークエンス解析)をおこない、原因遺伝子を探索しました(遺伝医療学分野との共同研究)。
従来法で診断確定できていなかった64名のうち新たに38名(59%)で症状と関連が疑われる原因遺伝子変異が確認されました。実際に、骨格筋組織でdysferlin蛋白の発現の低下が確認されている90名の横断的解析では、その70%にdysferlin遺伝子、10%にcalpain3遺伝子、5%に他の遺伝子の変異を認めていました。遺伝子診断率の向上が得られたとともに、dysferlin異常症を引き起こす遺伝子の種類やその割合などの具体的な遺伝学的背景が本研究により初めて明らかとなりました(9)。さらに、解析を継続することで、dysferlinの機能と密接に関わる分子を明らかにし、dysferlin異常症の筋細胞膜の修復障害機序を解明していきます。(井泉瑠美子)
図8. dysferlin異常症に見られる蛋白発現の異常.正常骨格筋組織で、筋細胞膜に発現しているdysferlin蛋白が、dysferlin異常症では発現していない. 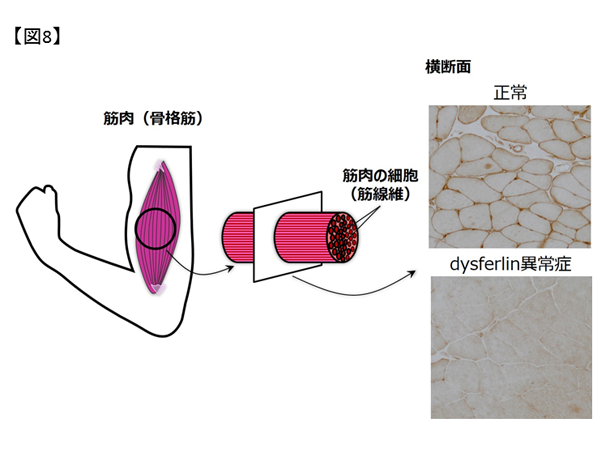
- 課題8. dysferlin異常症の病態解明と治療法開発
-
dysferlin異常症の病態解明と治療法開発 dysferlin蛋白は、筋細胞膜障害後の修復過程で重要な役割を持つ蛋白とされています。230 kDaもの大きな蛋白であるdysferlinは他の結合蛋白の機能を介して膜修復分子としてはたらく可能性があり、dysferlin結合蛋白の全容を明らかにするのはdysferlin異常症の病態解明や治療開発の上で重要と考えられます。
私達は新規dysferlin結合蛋白の同定を足掛かりとして、まだ全容が明らかでない筋細胞膜修復機構の解明とdysferlin異常症の治療法開発を目指しています。方法は下記の通りです。
(1) dysferlin蛋白の特定領域に着目し、アフィニティカラム法注と質量分析による新規結合蛋白の同定を行います(東北大学加齢医学研究所との共同研究)。
(2) dysferlin結合蛋白について、レーザー膜損傷の実験系を用いて患者由来細胞での細胞膜修復能を評価し、さらに薬剤スクリーニングにより細胞膜修復の治療候補探索を行います(香川大学・国際医療福祉大学との共同研究)。
(3) dysferlin異常症モデル動物(ゼブラフィッシュ、マウス)の運動機能や骨格筋病理の改善効果の検証を経て、治療応用につなげます(東京医科大学との共同研究)。
私たちは、(1)により複数のdysferlin結合蛋白を同定しました(図9.左)。同定した結合蛋白の一つであるProtein Xについて、培養細胞においてX遺伝子発現抑制が筋細胞膜修復機能の低下に繋がることを発見しました(図9.右)。さらに、X活性化剤の投与により培養細胞の膜修復機能が改善することを見出しています。今後は、X活性化剤について、モデル動物による検証を経て治療応用につなげたいと考えています。(小野洋也,鈴木直輝)
[注] ヒト細胞を使ったヒト蛋白質の複合体解析技術。細部内での分子数の少ない結合蛋白質を見つけ、ES細胞や神経、免疫細胞など種々の培養細胞での結合蛋白を同定出来るのみならず、特定の臓器細胞での相互作用蛋白を同定することや、膜蛋白や巨大複合体の結合を明らかにすることが可能。
図9. 左:複合体解析技術を用いたdysferlin結合蛋白の同定.右:レーザー膜損傷実験では、X遺伝子発現抑制により細胞外蛍光試薬が細胞内へ流入する. 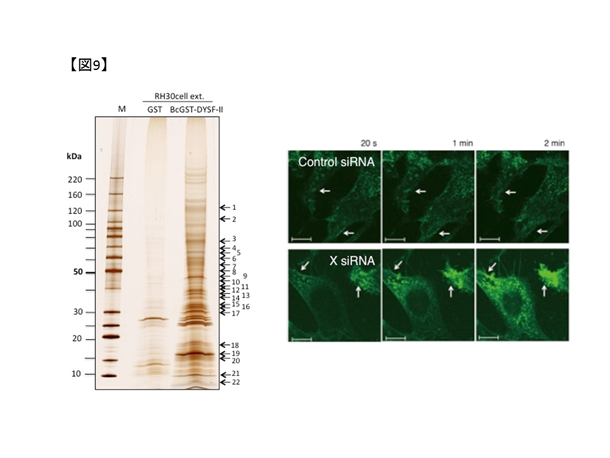
- 主な研究業績とリンク(ALS・筋チーム)
-
1. Mitsuzawa S, Akiyama T, Nishiyama A, et al. TARDBP p.G376D mutation, found in rapid progressive familial ALS, induces mislocalization of TDP-43. eNeurologicalSci 2018; doi: 10.1016/j.ensci.2018.04.001 2. Shijo T, Warita H, Suzuki N, et al. Aberrant astrocytic expression of chondroitin sulfate proteoglycan receptors in a rat model of amyotrophic lateral sclerosis. J Neurosci Res 2018; 96(2): 222-233. 3. Nishiyama A, Niihori T, Warita H, et al. Comprehensive targeted next-generation sequencing in Japanese familial amyotrophic lateral sclerosis. Neurobiol Aging 2017; 53: 194.e1-194.e8. 4. Suzuki N, Mori-Yoshimura M, Yamashita S, et al. Multicenter questionnaire survey for sporadic inclusion body myositis in Japan. Orphanet J Rare Dis 2016; 11: 146. 5. Nishiyama A, Warita H, Takahashi T, et al. Prominent sensory involvement in a case of familial amyotrophic lateral sclerosis carrying the L8V SOD1 mutation. Clin Neurol Neurosurg 2016; 150: 194-196. 6. Ichiyanagi N, Fujimori K, Yano M, et al. Establishment of in vitro FUS-associated familial amyotrophic lateral sclerosis model using human induced pluripotent stem cells. Stem Cell Reports 2016; 6(4): 496-510. 7. Akiyama T, Warita H, Kato M, et al. Genotype-phenotype relationships in familial amyotrophic lateral sclerosis with FUS/TLS mutations in Japan. Muscle Nerve 2016; 54(3): 398-404. 8. Burberry A, Suzuki N, Wang JY, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med 2016; 8: 347ra393. 9. Izumi R, Niihori T, Takahashi T, et al. Genetic profile for suspected dysferlinopathy identified by targeted next-generation sequencing. Neurol Genet 2015; 1(4): e36. 10. Izumi R, Warita H, Niihori T, et al. Isolated inclusion body myopathy caused by a multisystem proteinopathy-linked hnRNPA1 mutation. Neurol Genet 2015; 1(3): e23. 関連リンク(ALS・筋チーム)
https://www.researchgate.net/profile/Hitoshi_Warita
http://researchmap.jp/warita_2012
https://www.researchgate.net/profile/Naoki_Suzuki3
http://researchmap.jp/read0164712
- 「希少難治性筋疾患に関する調査研究」班
-
当教室では厚生労働省の難治性疾患政策研究事業の一つである「希少難治性筋疾患に関する調査研究班」の研究代表施設として患者数の少ない筋疾患を対象とし、疫学調査に基づく実態把握、診断基準・重症度分類の確立、診断ガイドライン等の整備を目標として、研究を続けています。研究分担施設も含めた対象疾患は、
・周期性四肢麻痺、非ジストロフィー性ミオトニー症候群といった筋チャネル病と多岐にわたります。当科では他の科学研究費等の支援をうけて、封入体筋炎・三好型ミオパチーは病態解析や治療開発、GNEミオパチーは臨床試験の側面でも注力しています。将来的な治療開発に向けての基盤整備を継続して続けていきたいと思います。
・先天性筋無力症候群
・Schwartz -Jampel症候群
・Danon病や過剰自己貪食を伴うX連鎖性ミオパチーなどの「自己貪食空胞性ミオパチー」
・封入体筋炎
・先天性ミオパチー
・縁取り空胞を伴う遠位型ミオパチー(GNEミオパチー)
・眼・咽頭遠位型ミオパチー
・三好型ミオパチー(およびその他の遠位型)
・マリネスコシェーグレン症候群
・べスレムミオパチー・ウルリッヒミオパチー
事務局連絡先:鈴木直輝 naoki*med.tohoku.ac.jp(*を@に置き換えてください)図:2016年度 班会議 
- 神経筋疾患の遺伝子解析
-
当教室では東北大学遺伝医療学分野、細胞増殖制御分野と協同し、神経筋疾患の遺伝子解析を行っています。対象疾患は、
・家族性筋萎縮性側索硬化症(家族性ALS)です。解析をご希望される方は事前に下記までご連絡ください。検体送付方法などをお知らせいたします。なお、遺伝子解析には患者さんご本人の文書による同意が必須で、主治医の先生方からの依頼のみ受けつけています。
・ジスフェルリン異常症を中心とした筋疾患
・その他、家族歴が明確な神経筋疾患(要ご相談)
(お引き受けしかねる場合もありますが、ご容赦ください)
連絡先:鈴木直輝 naoki*med.tohoku.ac.jp(*を@に置き換えてください)図:家族性ALSの遺伝子解析例(Suzuki N et al. J Hum Genet 2010より改変) 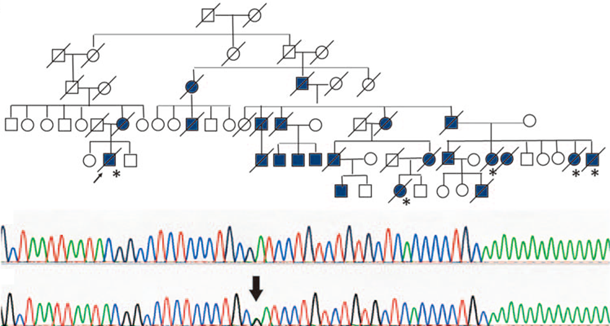
- 家族性ALS J-FASTレジストリ
-
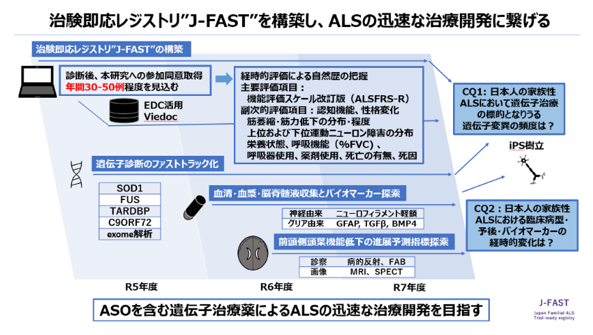
筋萎縮性側索硬化症(ALS)に対するアンチセンスオリゴ(ASO)による病的遺伝子産物の発現抑制治療開発が進んできています。レジストリの整備と迅速な遺伝子診断体制の強化により臨床病型・予後を明らかにすること、さらには治療効果判定に用いることのできる病態バイオマーカーを含めた自然歴の蓄積が急務となっています。
東北大学では日本医療研究開発機構AMEDの支援を受け、全国6施設と既存のALSレジストリJaCALSと連携し、家族性ALS患者を迅速にリストアップし経時的な病状の変化を把握する“Japan Familial ALS Trial-ready registry; J-FAST”を2023年度から運営しています。40歳以下の若年での発症または家族歴のあるALSの方がいらっしゃいましたら、研究へのご協力をよろしくお願いいたします。
*研究協力施設に通院中の患者さんへの依頼フォームはこちら。 - 骨格筋生検・末梢神経生検組織の病理学的解析
-
当教室では神経筋疾患の診断目的での生検筋・神経の病理学的解析を行っています。東北大学病院および連携医療機関の症例のみを対象としています。東北大学・医学系研究科の倫理委員会の承認も得た上で、残余検体を用いた病態解析研究も行っています。
図:眼咽頭遠位型ミオパチー(OPDM)に見られた縁取り空胞、赤色ぼろ線維およびcytoplasmic body (ゴモリトリクローム変法)の例 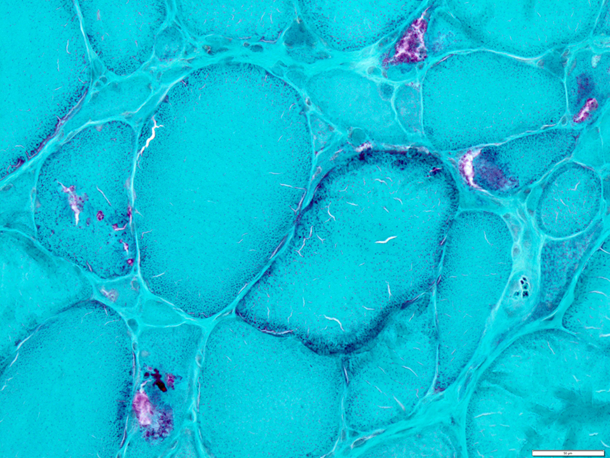
多発性硬化症・神経免疫疾患研究チーム(MSチーム)
三須建郎*,黒田 宙,西山修平,高井良樹,小川 諒,金子仁彦,小野紘彦;伊藤香蘭(技術補佐員),熱海万裕(研究支援者)
大学院入学を希望される皆さんへ

わたしたちMSチームは、従来の脱髄疾患の概念を覆す数々の研究成果を世に送り出してきましたが、その基本はいつも患者さんを診ることで、ベッドサイドから生まれた研究ばかりです。あわせて基礎的研究も行いつつ、国内外の研究者と共同研究を推進し、みなで和気藹々と研究を行っています。ベッドサイドでの「なぜ?」に答えることに重きを置いており、実際に世界的な研究の多くは大学院生の手で行われてきました。私達と一緒に世界にインパクトを与えるような研究に携わってみませんか?
MOG抗体の測定依頼についてはこちらへ
筋萎縮性側索硬化症・骨格筋疾患研究チーム(ALS・筋チーム)
割田 仁*,鈴木直輝,井泉瑠美子,西山亜由美,小野洋也,秋山徹也,池田謙輔,四條友望,光澤志緒;島倉奈緒子(技能補佐員),待井明子(技術補佐員)
大学院入学を希望される皆さんへ

難治性神経筋疾患にとりくむ私たち研究チームは、”研究者に上下なし”をモットーに人の和を大切にしつつ、お互いに励まし合って研究を進めています。大学院生となられた方達には、基本的実験手技はもちろん研究開始に必要な知識を万遍なく伝授する「基本コース」(半年間)を用意し、1〜2週に一度のミーティングでは文献の読み方からプレゼンテーション技法まで、皆さんの求める大学院教育に力を入れています。さぁ、私たちと一緒に研究を始めてみませんか。
現在、私たちは次世代シークエンサーによる網羅的遺伝子解析、iPS細胞・ゲノム編集技術を用いたヒト細胞モデルの開発、動物モデルを用いた治療法開発研究をおこなっています。当教室の橋渡し研究(TR)実践を代表する2つの医師主導治験は現在まさに進行中です。
![]()